
DML Analysis Part 16
Mox update
I stopped bismark job I started on Mox because I didn’t specify a path for samtools. Steven figured this out manually because by redirecting my standard error, there was nothing going into my default slurm output from Mox. I edited my script to include a path to samtools and remoe redirecting any error. I then restarted my Mox job. It’s currently deduplicating my files, but I used cat slurm-401682.out | grep "Mapping efficiency *" to look at the mapping efficiencies for the alignment.
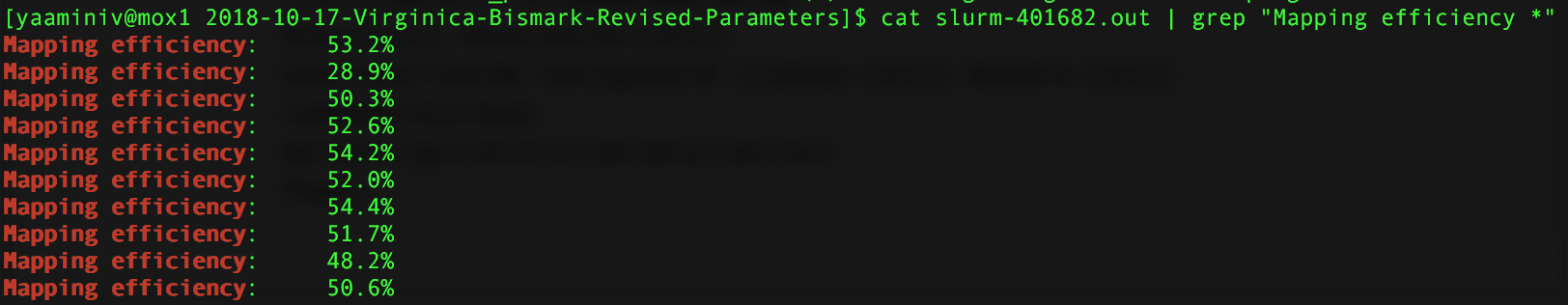
Figure 1. Mapping efficiency for Mox alignment. The first mapping efficiency is for sample 10, second-tenth for samples 1-9.
Even though my Mox alignment used Bowtie 2-2.3.4, and my genefish alignment used Bowtie 2-2.2.9, I got the same mapping efficiencies! I created a table in my paper draft that Steven looked at. Turns out he was getting different mapping efficiences because he did not specify that the input data was --non_directional. I verfied that I needed that argument in this issue. The reason why Steven was getting different mapping efficiencies was because he did not include this argument.
However, I encountered another problem with my revised Mox run: I didn’t specify --samtools_path in my alignment step! I didn’t need to do this on genefish because samtools was already in my computer path. Because Mox couldn’t find samtools my output files are now SAMfiles.
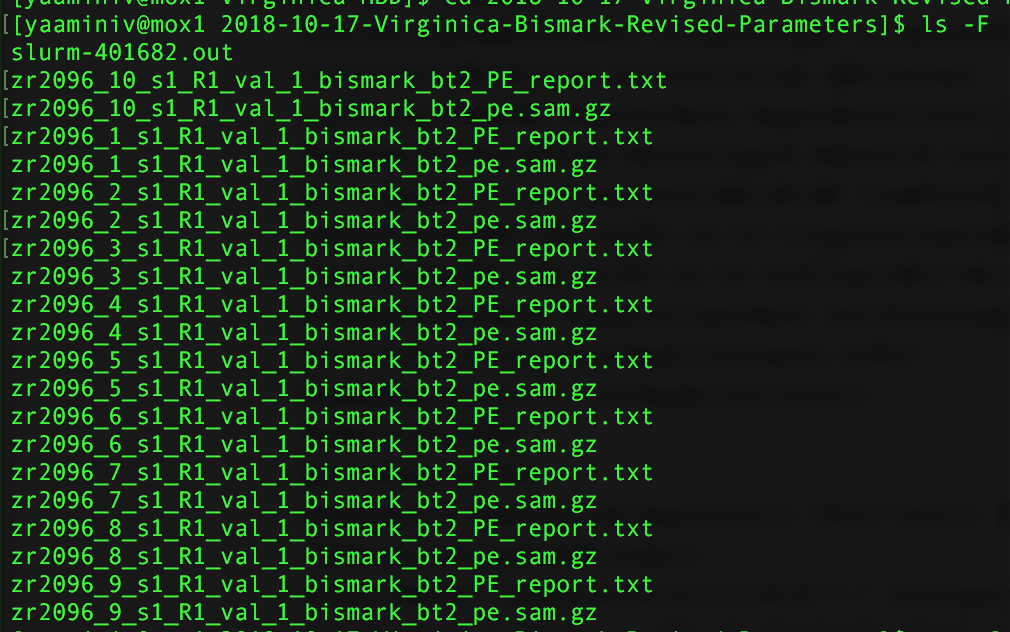
Figure 2. Output from bismark alignment.
I need to convert my SAMfiles to BAMfiles before I move onto methylKit. I also added --samtools_path in a new script and queued the job on Mox.
I then encountered a THIRD issue with Mox. Just looking at my slurm.out, I couldn’t tell if my deduplication was actually running, or if it encountered an error.

Figure 3. Status of slurm.out file
I posted this issue. Sam pointed out that my script included a “" at the end of the last line of code, which essentially left it hanging. I edited this script to remove any hanging backslashes, cancelled both of my queued jobs (Sam later mentioned that I probably should not have cancelled both but oh well), and restarted a new Mox job.
From now onwards, I need to quadruple check all of my Mox scripts. Because I’m not running the bismark pipeline in chunks like I’m used to in Jupyter, it’s hard to catch errors, fix them, and easily restart from where I left off.
Going forward
- Figure out how to convert SAMfiles to BAMfiles
- Use
methylKitto identify DML and DMR - Characterize DML and DMR locations