
DML Analysis Part 28
Revisiting DML analyses
Perfecting the DML track
Based on this issue, I revised the destranded 5x DML track to show methylation differences instead of a false forward strand indiciation. Turns out I applied mutate on the wrong dataframe in my R Markdown script! My revised track, found here, matched the track Steven generated.

Figure 1. DML tracks in IGV with 5x and 10x coverage sample tracks.
This is also interesting because Steven used filterByCoverage after processBismarkAln to generate the 5x coverage track, while I used mincov within processBismarkAln. It is interseting to know that we (presumably) get the same DML locations. I decided to go forward with the destranded 5x DML track, since both Claire and Mac used 5x coverage for their analyses.
CpG information
I returnedd to my R Markdown script and got CpG coverage and methylation information for the 5x processed samples.
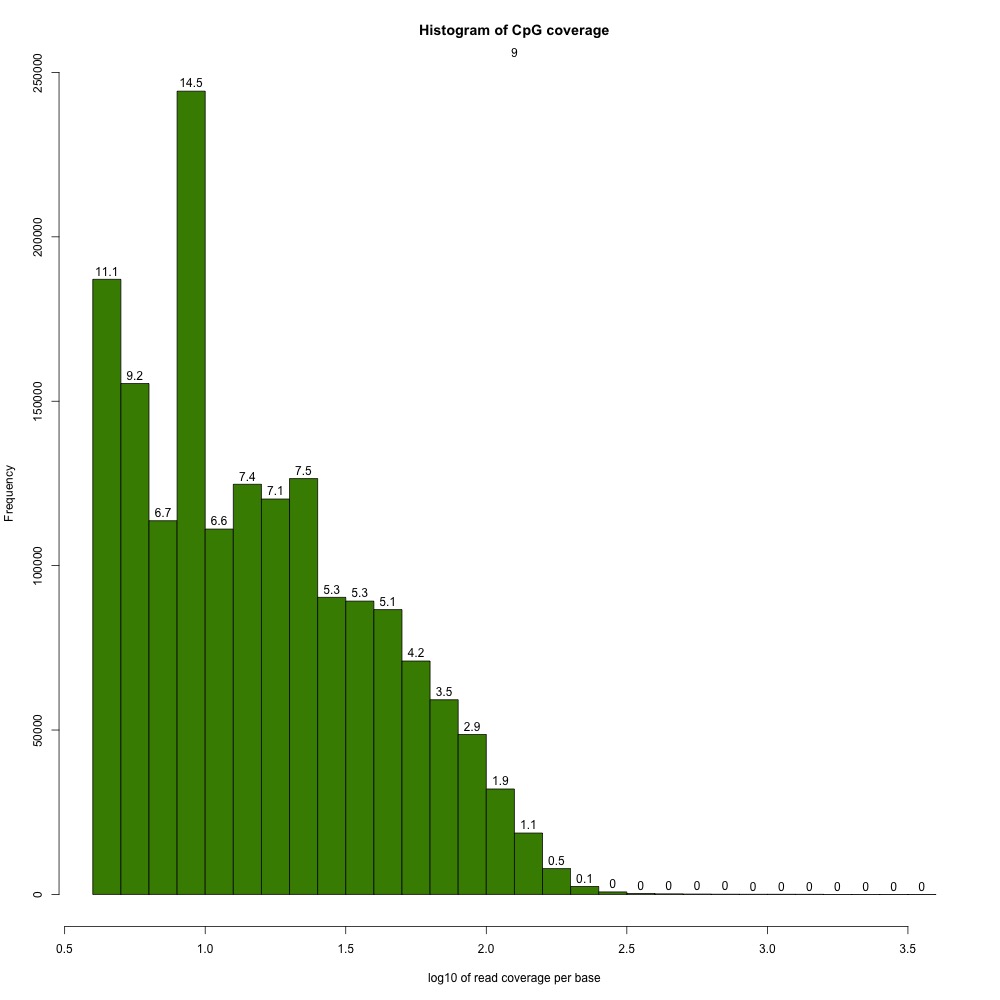
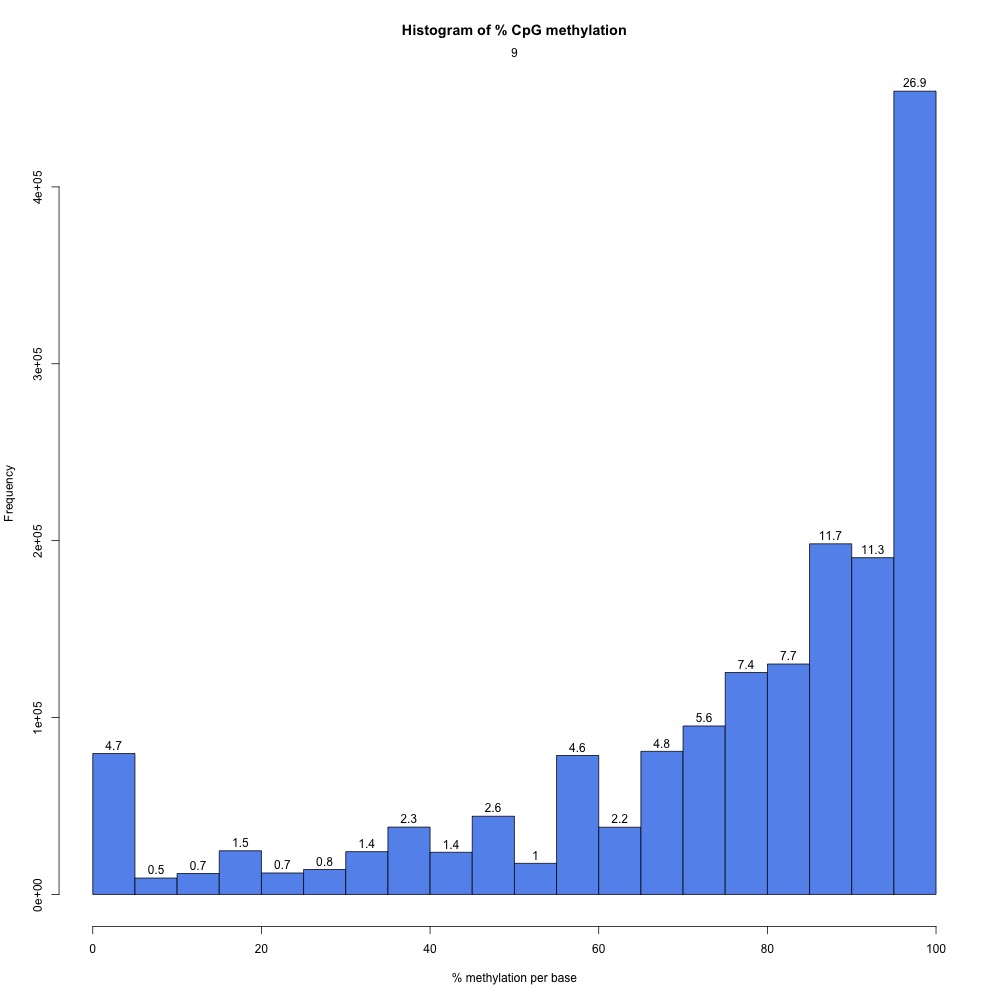
Figures 2-3. Example CpG coverage and percent methylation plots.
I also generated a correlation plots, full sample methylation clustering, PCA, and screeplots within methylKit.
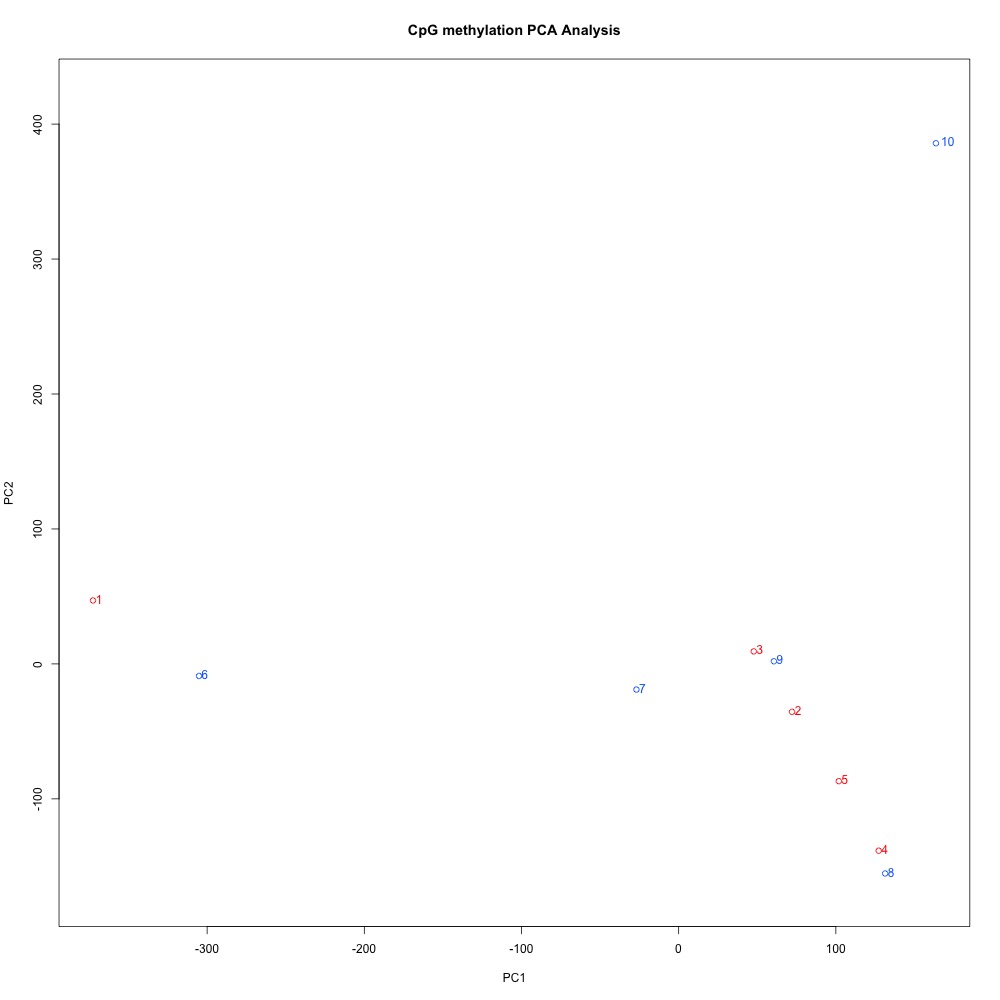
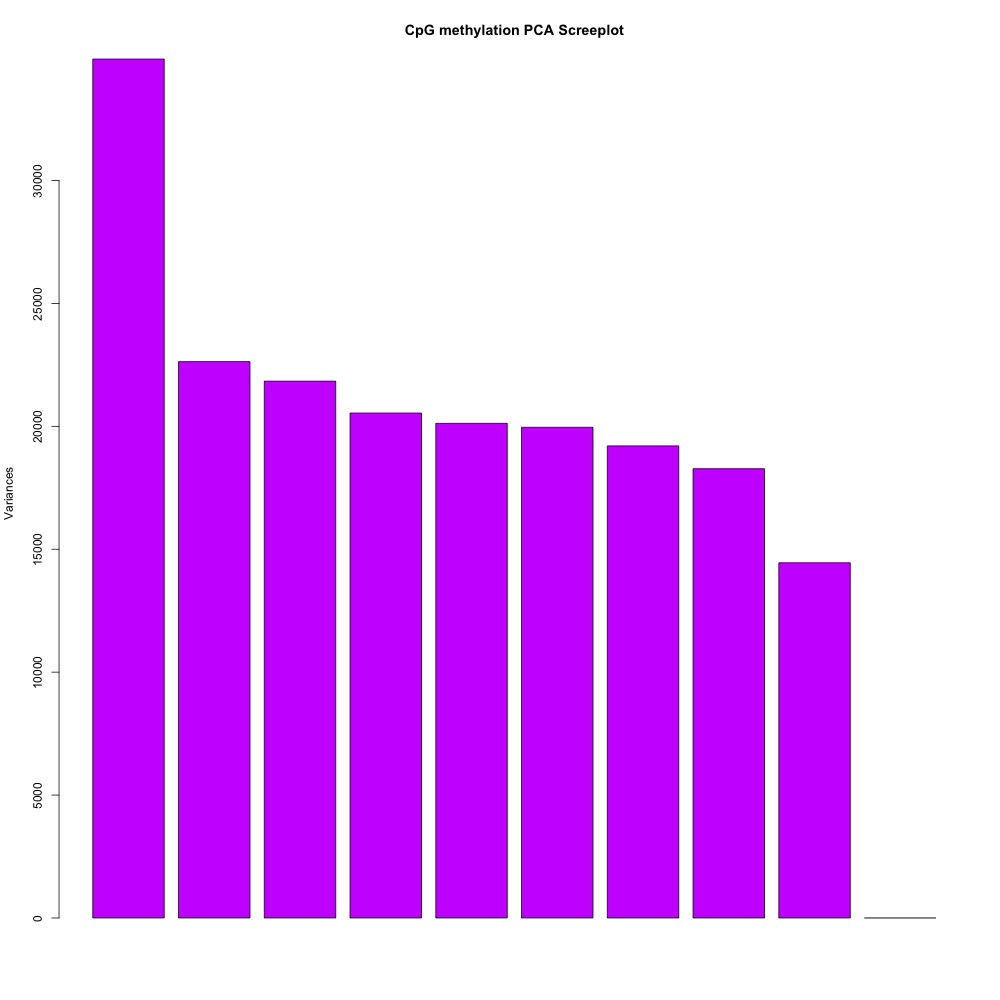
Figures 4-7. Full sample plots.
Characterizing DML locations
I revisited my bedtools Jupyter notebook to characterize the location of destranded 5x DML. I reset the variable path name (side note: HOLY FORK SUCH A HANDY TOOL I’M SO GLAD I USED IT) to the new destranded 5x track. I then went through the script and reran all of my DML code. All of the overlap locations can be found here. I also reran my flankBed and closestBed analysis, and saved the output here.
The track had 630 DML, instead of the 1398 in my 3x coverage track. Of the 630 DML, were 335 hypermethylated in the treatment and were 295 hypomethylated. Most of the DML, 576, were located in mRNA coding (genic) regions, with 1474 genes represented. In the genic regions, 388 DML overlapped with exons and 200 overlapped with introns. 61 DML overlapped with transposable elements generated using all species in the database, and 40 DML overlapped with transposable elements generated using C. gigas only. In 1000 bp flanking regions upstream and downstream of mRNA coding regions, 46 DML overlapped with upstream flanks and 50 overlapped with downstream flanks.
Edit 2019-03-17:
Steven suggested I count the number of DML that do not overlap with any features (i.e. are in unannotated intergenic regions). To do this, I used the -v argument in intersectBed, which reports “those entries in A that have no overlap in B. I also specified multiple files with -b: exons, introns, and transposable elements identified using C. gigas only. I don’t need to include mRNA coding regions, since exons and introns are included in that file. I found that 39 DML did not overlap with my feature tracks. I saved the list of DML here.
Going forward
- Conduct a proportion test for DML locations
- Describe methylation irrespective of treatment
- Update paper methods and results
- Work through gene-level analysis
- Figure out what’s going on with DMR
- Figure out what’s going on with the gene background