
Gonad Methylation Analysis Part 13
The whole enchilada (but better this time)
Now that I’ve tested the entire bismark to methylKit pipeline on a 10,000 read subset for each C. virginica sequencing sample, I can begin validating the full pipeline. Steven already ran the full samples on this pipeline in this notebook. My job is to reproduce his workflow.
I opened my full pipeline Jupyter notebook. Since I’m using trimmed files from this directory, the first thing I wanted to do was test my find and xargs commands to ensure I isolated the right files.

Figure 1. Testing find and xargs on a new set of filenames.
Then, I piped the find and xargs output into bismark. Unlike Steven, I did not set a parameter for -score_min. In our conversation today, we decided that I should run the alignment with the default parameter so we can compare our results. The default is L,0,-0.2 (instead of L,0,-1.2 used by Steven).
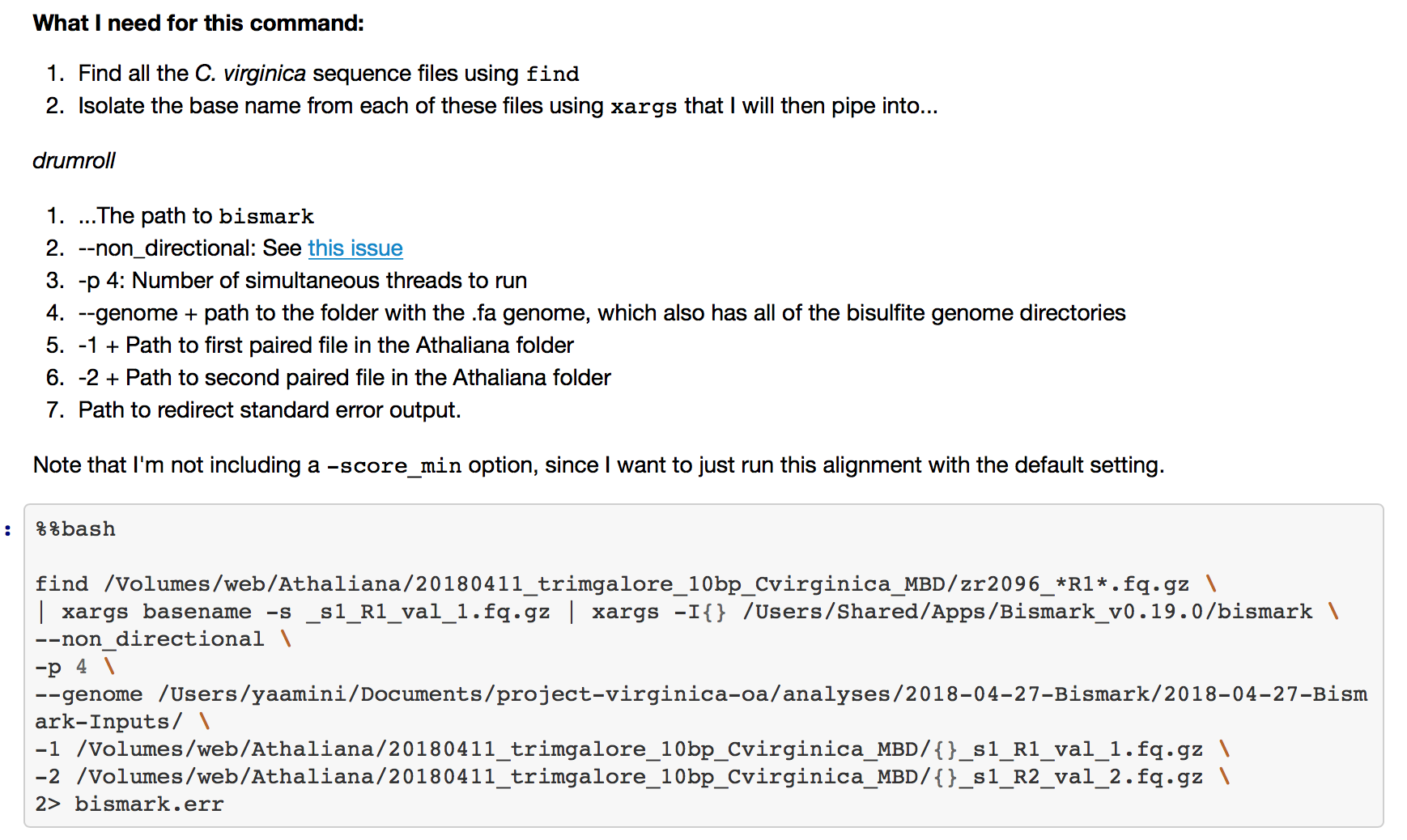
Figure 2. Description of each line of code, along with the actual command.
It took me a bit of finnagling to get that code to work because of some typos! I’m going to jot down a few reminders for myself:
- Always check to see if each line has a “"
- Use tab complete to ensure lack of typos and the correct path to files
- Verify you’re using the correct comand
- Test small bits of code before putting everything together
I’ll check on the alignment progress tomorrow, but my guess is that this will take 2-3 days to complete. Once the alignment is finished, I will deduplicate, sort, and index the resulting .bam files.