
MethCompare Part 2
More genome feature tracks
After I looked over code and added analyses, approaches, and figures we could explore in the paper, I set out to refine genome feature tracks for M. capitata and P. acuta. Once we confirm we trust our analysis files, I can use these feature tracks to understand where methylation occurs in these species.
M. capitata CDS track
The first thing I did was examine the M. capitata CDS track further. I went back to this Jupyter notebook where I generated all the genome feature tracks. The first thing I realized was that I didn’t make the gene track correctly! When I used grep "gene", it pulled lines with CDS and intron information and saved that to the gene track. I used grep AUGUSTUS gene" instead, and used similar code for the rest of the M. capitata tracks.
Now to the task at hand: understanding the CDS track. Looking at the gene track using head, I could see that CDS were split up by introns. In that way, the CDS track is similar to an exon track.
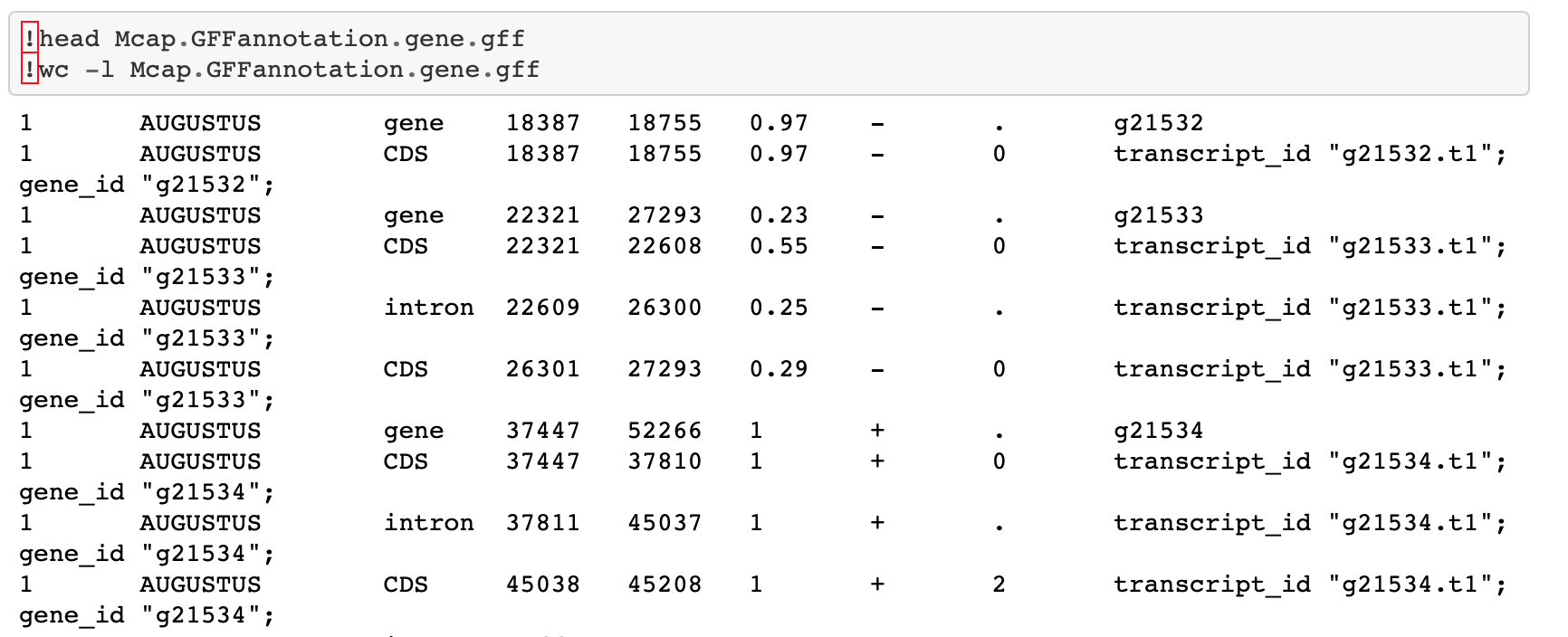
But looking at all the tracks in this IGV session, I don’t think the CDS track includes UTR.

I posted this issue to get more clarity about the CDS track and see if we can derive UTR or exon information from the gene and CDS tracks. If not, then I don’t think I’ll be able to do comparisons between M. capitata and P. acuta.
Visualizing P. acuta tracks
The P. acuta tracks were in better shape: I have gene, transcript, exon, intron, and CDS information. Within exons, I have initial, internal, and terminal exon tracks that can help us answer a lot of questions about exon-specific methylation. I wanted to create an IGV session for all the P. acuta tracks. I downloaded the genome from the Google Drive and tried visualizing the intron track I generated in this notebook. In IGV, this track looks blank (even though I know from my files that there are introns on this scaffold):


I posted this issue to get some help. I thought maybe the file wasn’t sorted correctly, but since I pulled it directly from the genome I don’t think that would be the issue. We’ll see.
Going forward
- Create promoter, UTR, and intergenic region tracks for species depending on what information is available and what is possible
- Intersect all genome feature tracks with CG motif information
- Rerun the CpG characterization pipeline with full samples and incorporate new genome features
- Create concatenation files and figure out methylation island analysis