
WGBS Analysis Part 37
Thinking about SNPs
Steven and I have gone through the BS-Snper code and results multiple times this week to determine how to deal with SNPs that overlapped with DML. We dove into the BS-Snper program information to understand how it could identify C->T SNPs over bisulfite-converted unmethylated CpGs. During bisulfite conversion, unmethylated cytosines are replaced with uracils, then converted to adenines during PCR amplification. The complementary strand for this locus would have a guanine, since the original base pair was a cytosine. If there’s a C->T SNP, then the complementary strand would have an adenine.
Once we clarified the methods, we realized that the SNPs needed to be excluded from any DML characterization: they are false DML! I removed the SNPs from DML using subtractBed in this script. I used my revised DML track with no SNPs to characterize genomic locations. Once I updated the counts, I returned to this R Markdown script to revise my heatmap and chromosome distribution figures. Then, I used the updated counts in this R Markdown script to update my stacked barplot and gene count table.

Figure 1. Heatmap of unique DML

Figure 2. DML distribution over chromosomes
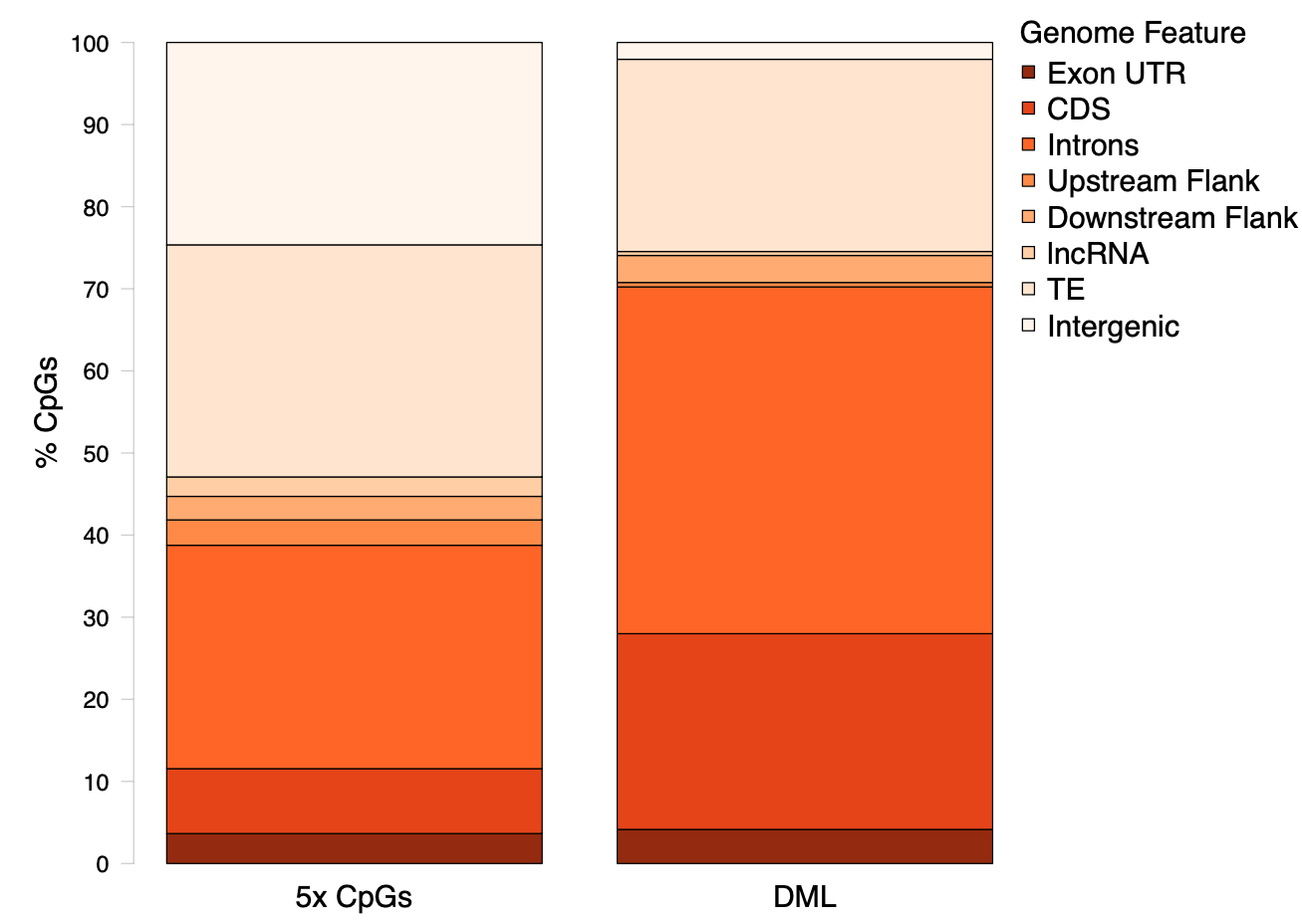
Figure 3. Genomic location of DML
I also went through and removed SNPs from my general methylation characterization. No point characterizing the methylation status of loci that are incorrectly on that list anyways! I used this Jupyter notebook to remove SNPs from the 5x union BEDgraphs I used to separate highly, moderately, and lowly methylated CpGs. I used the updated counts in this R Markdown script to revise my figures and supplementary tables.

Figure 4. Genomic location of CpG categories
Nothing left to do now but put the finishing touches on!
Going forward
- Revise title
- Format for submission
- Submit preprint to bioRXiv
- Submit paper for publication
- Report
mc.coresissue tomethylKit - Perform randomization test
- Determine if larval DNA/RNA should be extracted