
WGBS Analysis Part 4
Looking at preliminary DML
I have DML!. To clarify, I have 15,386 DML when filtering my data for 5x coverage. That’s more than twice as many DML as I got for my C. virginica data. Before I characterize the location of my DML in the C. gigas genome, I’m going to do two different things…
Trying different coverage in methylKit
Since I used WGBS, there’s a chance that I can use a more stringent coverage filter instead of 5x coverage. I returned to my R Markdown script and reprocessed the data using 10x coverage. I somehow ended up with more DML using 10x coverage! The files I generated can be found below:
Looks like the reason why I have more DML when using 10x coverage is because I gained 300 hypermethylated and 600 hypomethylated DML. The increase in hypomethylated DML is interesting. My C. virginica data had a pretty even split of hyper- and hypomethylated DML, whereas with 5x coverage I have 700 more hypomethylated DML and 1,000 more hypomethylated DML with 10x coverage. I don’t entirely know why this is happening so good thing I already planned on checking things out in IGV.
Table 1. Number of CpG loci in various categories
| Coverage | DML Background | All DML | Hypermethylated DML | Hypomethylated DML |
|---|---|---|---|---|
| 5 | 11,285,932 | 15,385 | 7,384 | 8,001 |
| 10 | 5,210,744 | 16,324 | 7,669 | 8,665 |
Verifying DML in IGV
I need to confirm that my DML are real, and the best way to do that is in IGV. I realized my IGV veresion was outdated, so I downloaded version 2.6.3 from the website. Since I’m looking at different coverage metrics, I also wanted to generate some bedgraphs that matched the coverage settinsg I used. I created this Jupyter notebook by copying my C. virginica equivalent and created 5x and 10x begraphs for each sample. After moving the contents of my repository to gannet, I used URLs to add the following tracks to a new IGV session:
- C. gigas genome
- YRVA 5x bedgraph
- YRVA 10x bedgraph
- YRVL 5x bedgraph
- YRVL 10x bedgraph
- DML background 5x coverage
- DML background 10x coverage
- DML 5x coverage
- DML 10x coverage
I actually struggled quite a bit to load all of the tracks into the IGV session. I made the mistake of adding the full bedgraphs in the IGV, but couldn’t open the file to delete the tracks. When I tried editing the xml file itself, I could successfully add tracks but not remove them. I ended up deleting my session and starting a new one. Once I created a new session, I could do what I needed!
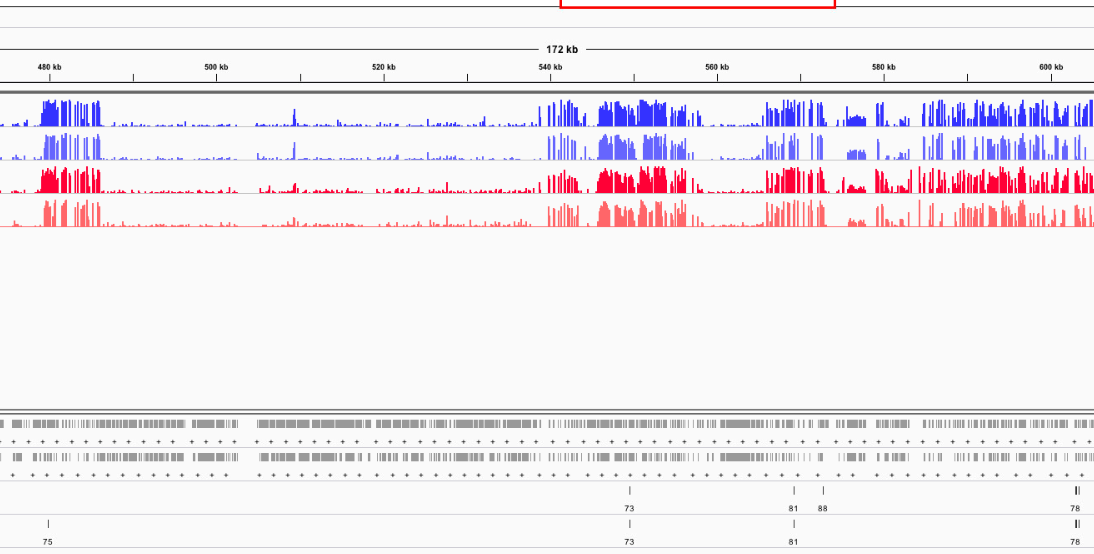
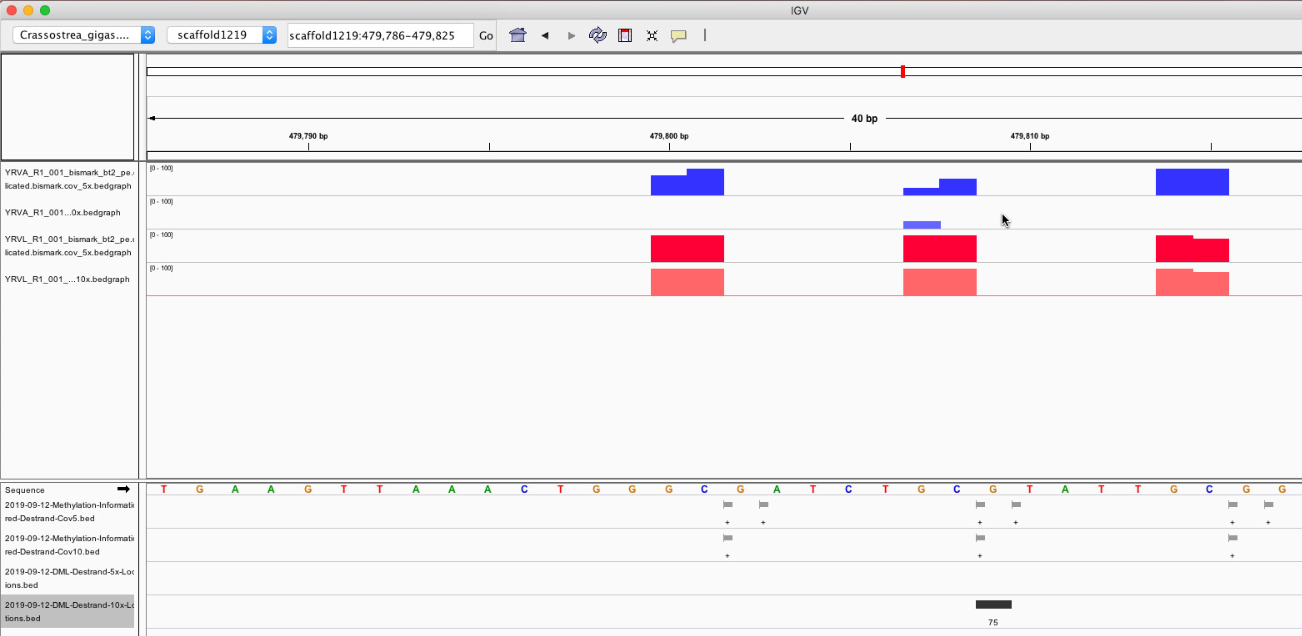




Figures 1-6. DML on various chromosomes.
Looking at the DML, I can see that I didn’t export my BEDfiles correctly. The DML background and DML themselves don’t overlap neatly with CpGs (they’re 1 bp off). I need to change how I modify the base pairs in methylKit and resave the tracks.
I also don’t trust the 10x DML that were not in 5x DML. I don’t know what’s going on, but it’s flaming hot garbage. The other DML in the 10x track that were common to the 5x track looked good, so I think I want to remove all 10x DML that are not common to the 5x track. I will figure out a way to do that.
Quick bismark update
The alignments went well! My first file deduplicated successfully, but my second file was saved with the wrong filename. I realized my wildcard was not specific enough, so I created a modified script from deduplication onwards and saved it here. I moved the script to Mox and started the job. Once I get the files reprocssed I’ll not only run through my analyses, but also create one master script so I don’t have to piecemeal anymore.
Going forward
- Get revised
bismarkoutput from Mox and run through analysis pipeline - Fix DML background and DML BEDfiles in
methylKit - Remove 10x DML that are not present in 5x DML track
- Obtain C. gigas feature tracks
- Characterize locations of DML
- Conduct a gene enrichment and/or identify genes with DML and discern what it means