
MethCompare Part 3
Finalizing genome feature tracks
Based on feedback to only generate tracks common between species, I focused on validating the tracks I generated already for M. capitata and P. acuta.
M. capitata tracks
Since M. capitata does not have any transcript, exon, or UTR information, I can’t generate UTR or promoter tracks. While I could generate an intergenic region track, I figured this might be redundant. With intersectBed -v, I can figure out what CpG loci intersect with intergenic regions using the gene track. If Hollie wanted to make Circos plots with various gene tracks, any locus that falls outside of a gene would be considered intergenic. I held off on making these tracks for now, but I may revist later if we decide they are necessary.
Earlier this week, I posted this issue asking for CG motif tracks for both species. I used the links from that issue to add CG motifs to this IGV session with M. capitata feature tracks. Hopefully this will be useful for us later.
P. acuta tracks
I wasn’t able to visualize the P. acuta tracks I generated in IGV, so I posted this issue. Steven pointed out that the scaffold names did not match the names in the original genome file, since there was an additional underscore in scaffold names in the genome file but not in the genome feature tracks. I posited a complicated plan to add this underscore to the right place, but Sam usggested I streamline my multistep process by using sed.

Using these modified tracks, I was able to see them in IGV! Even though P. acuta has separate CDS and exon tracks, they’re made up of the same components. It looks like the CDS are transcript specific and string together what is actually coded for each transcript, while thye exons are split up into initial, internal, and terminal sequences that may shed more light on what specific sections go into each transcript. Looking at the gene, transcript, exon, and CDS start positions, I did not see any UTR. That is consistent between species, so I wouldn’t have been able to generate that information to begin with.
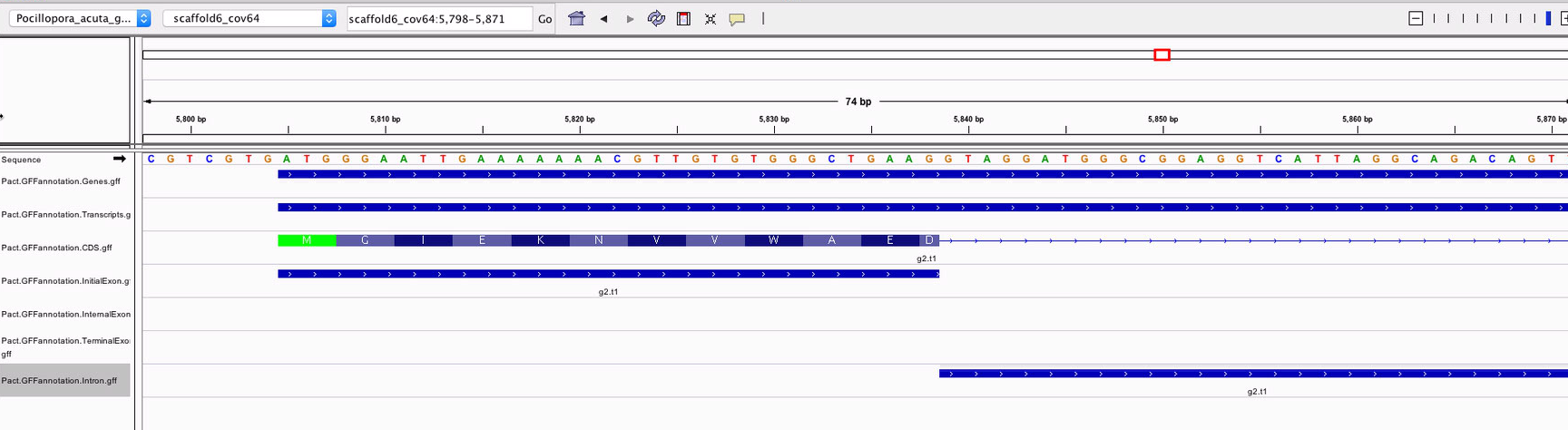
When spot-checking the tracks to make sure they were valid, I noticed that the introns seemed to overlap with coding sequences.
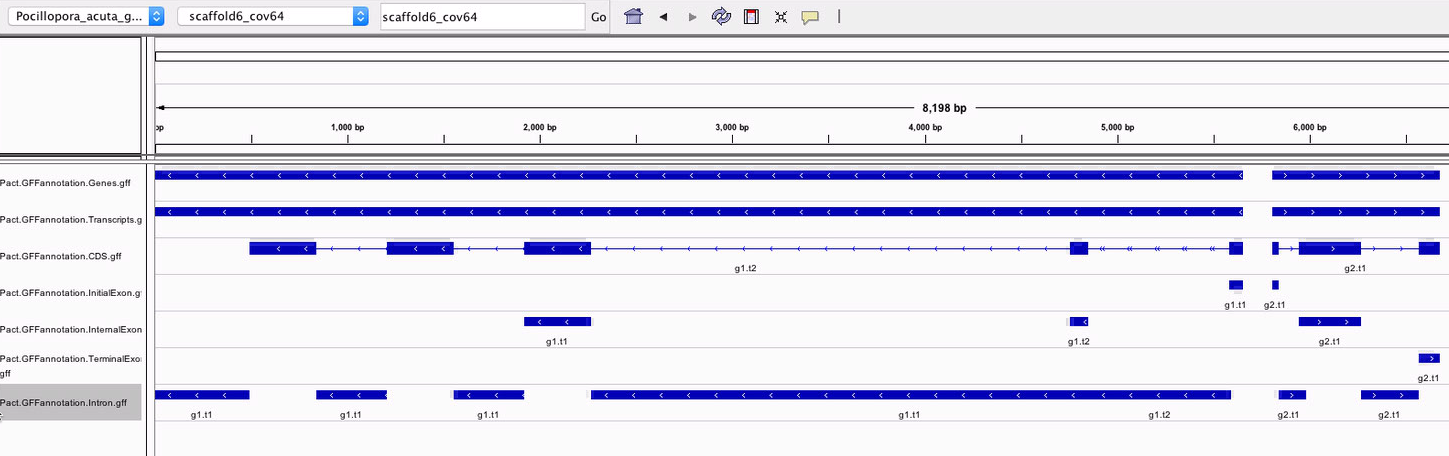
Looking closer, the transcript-specific introns were being mashed together, instead of showing two overlapping features.

Since I know they are separate in the actual tracks, it wasn’t the end of the world, but it was frustrating that I wasn’t able to visualize it properly. I posted this issue to see if I can get them to display as separate features. Sam suggested I look at the original GFF I pulled the tracks from, but this would involve adding the underscore to scaffold names after pulling out all AUGUSTUS lines. He also suspected that this would be an issue regardless becuase coordinate-based systems like IGV probably merged overlapping features. He was corret. When I looked at the CG motifs, I found that two adjacent motifs were combined into one feature.

While it’s not ideal, I know that it’s probably not my fault. I added the CG motifs to this IGV session for P. acuta tracks and decided to move on.
Going forward
- Intersect all genome feature tracks with CG motif information
- Set up CpG characterization pipeline with revised tracks
- Rerun the pipeline with full samples
- Create concatenation files and figure out methylation island analysis