
WGBS Analysis Part 5
Finalizing the analysis pipeline
I’m still waiting on final bismark output, so I figured I’d finalize my analysis pipeline and start working on a draft talk. I should have my final files tomorrow, which will allow me to make any tweaks before my practice talk Monday!
More bismark script issues
I woke up this morning and saw my Mox job completed! When I checked my file list, I saw that my low pH sample had no deduplicated or sorted file. Instead, the script created 2 different ambient samples. Frustrated with wildcards, I modified this script to spell out each file I wanted processed. I ran the script and peeked at the output — my low pH sample is being processed correctly! When I make my master script, I need to include the specific wildcard for one character (I think it’s ?) to ensure all my files will be processed and the output will be named properly.
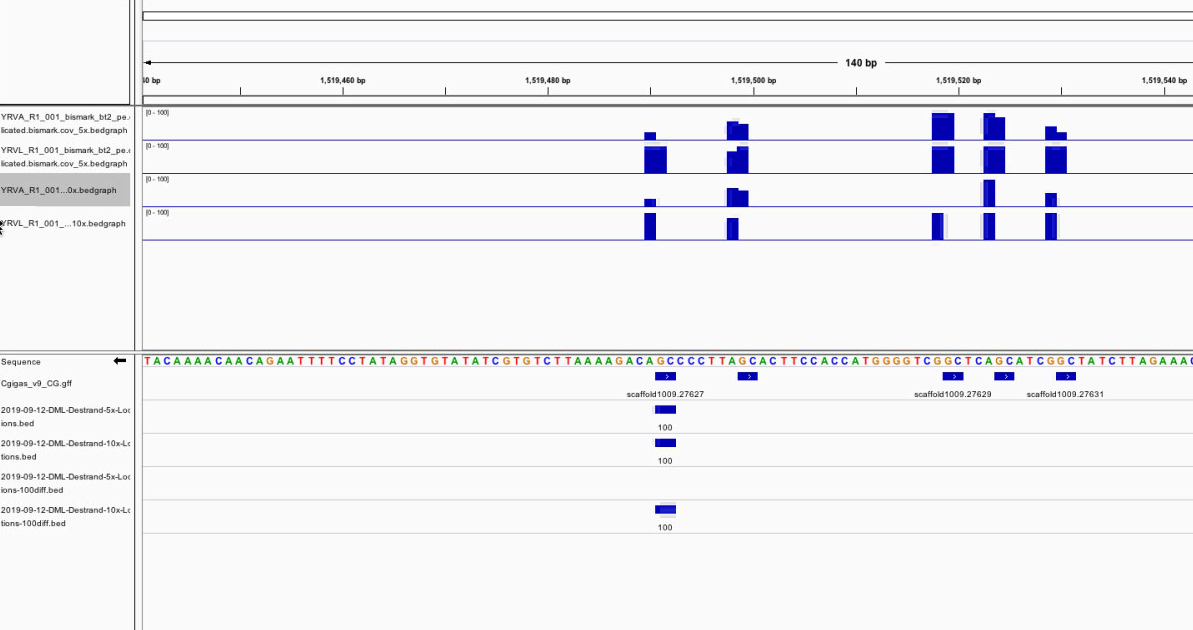
Checking start and end positions for IGV
Yesterday I noticed that my bedgraphs, DML background, and DML locations weren’t aligning. I tried shifting the DML background up one position but that didn’t line up either. Looking at my C. virginica IGV screenshots I noticed a similar visual discrepancy for bedgraphs vs. my other tracks. I decided to load the C. gigas CG motif track to compare my tracks to something that’s already been tested.

Figure 1. CG motif track with other DML and DML background tracks.
They don’t line up! I definitely need to move the loci start position down 2 and end position down 1. I was able to fix that just fine. However, I noticed that the root problem may be that the unite output has the start and end positions of the switched and potentially switched.

Figure 2. unite output.
If I use destrand = FALSE, I don’t get that problem.

Figure 3. unite output with destrand = FALSE.
I guess I have a lot more data on the reverse strand than I expected. I know I need to destrand, but perhaps I don’t need to modify base pair positions for the DML background. There’s also a chance that loading in finalized data from my Mox run will change some things. I’ll stick to just getting my loci to look good and worry about the DML background after PCSGA.
Changing getMethylDiff parameters
Since I was getting over 15,000 DML, Steven suggested I change the stringency of gethMethylDiff parameters. This made sense because I am only working with two different samples, and I want to ensure my results are “real.” With the 5x coverage data, I tried setting my methylation difference minimum to 75% and 99% in this R Markdown script. I used 99% since using 100% difference gave me an error, but still only returned loci that were 100% between samples. Using a 100% difference between samples, I still had ~2,000 DML. I then made my q-value stricter, going from 0.01 to 0.001. These settings gave me ~800 DML, which I felt was reasonable for WGBS. I used 100% difference and q-value < 0.001 with the 10x coverage data as well. This time, I had less DML with 10x coverage data! I links to the files I saved can be found below:
5x coverage:
10x coverage:
Verifying DML in IGV
I was still unsure if there are some DML that are more artifacts of losing coverage, so I went back to IGV.


Figures 4-6. Example of 10x DML that does not exist in 5x data.
Looking at the DML above, it looks like it was not included for the 5x coverage data since there was missing data at 5x coverage, but the 10x coverage data was there for both samples (no second lump for 10x data, second lump there for 5x low pH data but not ambient pH data). The second locus, however, makes no sense to me. There loci isn’t there in the 10x coverage data, so why is it a DML?! Then there’s the third locus. Again, doesn’t make sense to have a 10x 100% diff DML there, but it also doesn’t align with the CG motif. This visualization is going to be difficult, but at least it overlaps with the CG motif so I can still use bedtools.
Cleaning up DML
I want to remove the any DML in the 10x 100% difference data that are not in the 5x 100% difference data. I figured I could do this with bedtools by extracting the DML that are in both datasets. I don’t think it makes sense to have a 10x DML that is not in the 5x data, so this allows me to remove any extra 10x DML. At the end of this R Markdown script I used intersectBed to remove extraneous DML:
#Path to intersectBed
#Print line a information for overlaps
#File a: 10x 100% diff DML
#File b: 5x 100% diff DML
#Output filename
/Users/Shared/bioinformatics/bedtools2/bin/intersectBed \
-wa \
-a 2019-09-12-DML-Destrand-10x-Locations-100diff.bed \
-b 2019-09-12-DML-Destrand-5x-Locations-100diff.bed \
> 2019-09-12-DML-Destrand-10x-Locations-100diff-NoExtras.bed

Figure 7. Looking at the new track in IGV
Succss! The code worked. The files can be found here
Going forward
- Get revised
bismarkoutput from Mox and run through analysis pipeline - Obtain C. gigas feature tracks
- Characterize locations of DML
- Conduct a gene enrichment and/or identify genes with DML and discern what it means